
NL Journal of Dentistry and Oral Sciences
(ISSN: 3049-1053)
Oral Manifestations of Goltz Syndrome – A Case Report
Author(s) : Boudi Rachid, Ramdi Hind, Majid Sakout, Elalloussi Mustapha. DOI : 10.71168/NDO.02.03.116
Abstract
GOLTZ syndrome, also known as focal dermal hypoplasia, is a rare disorder characterized by specific clinical features. The patient with this syndrome has a macrostomy, underdeveloped maxillary and mandibular bones, and a high, hollow palatal vault. Early prevention and regular follow-up are essential to manage dental abnormalities and severe polycavities. Keywords: Goltz syndrome, rare, dental manifestations
Introduction
Goltz syndrome (or focal dermal hypoplasia) is a rare disorder affecting the skin, bones, and connective tissue. It is characterized by polymorphic skin involvement and various abnormalities that can affect the eyes, teeth, skeletal system, central nervous system, as well as the urinary, gastrointestinal, and cardiovascular systems [1,2].
This syndrome is most often identified in the neonatal period when it presents as a significant polydystrophic form; however, many milder cases may go undiagnosed. The prevalence is currently unknown [2,3].
The objective of this work is to present a clinical case highlighting the general and oral manifestations of this syndrome. This could aid in improving diagnostic and therapeutic approaches for dentists.
Clinical Case
A 3-year-old female from consanguineous parents, had a normal pregnancy and full-term birth. She is presenting with Goltz syndrome and has been referred by her pediatrician to the pediatric prevention department at the Faculty of Dentistry in Rabat for oral cavity rehabilitation.
Clinical examination reveals multiple signs: global hypotrophy relative to age, right facial asymmetry, right ectrodactyly, bilateral syndactyly of the 2nd and 3rd toes, areas of pink and achromic systematized hypoplasia with molluscoid lesions, and perioral pseudo-papillomatous appearance. The patient is under cardiology care for single ventricle, pulmonary artery stenosis, and tricuspid atresia. The patient presents a cleft lip , palatal archwich is elevated and concave and polycavities (figures: 1,2,3,4,5,6,7,8,9,10).
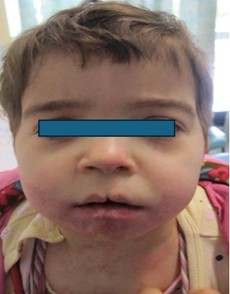 Figure 1: General signs
Figure 1: General signs
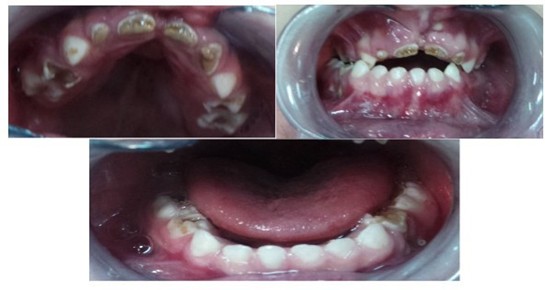 Figure 2,3,4: Endooral view showing severe polycavities
Figure 2,3,4: Endooral view showing severe polycavities
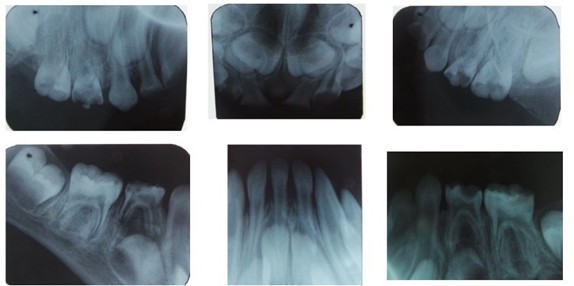
Figure 2,3,4,5,6,7,8,9,10: X-ray Assessment
Discussion
GOLTZ syndrome, also known as focal dermal hypoplasia, is a rare disorder characterized by specific clinical features. These include areas of skin atrophy and periorificial papillomas, predominantly around the mouth, genital, and anal regions. Hernias of subcutaneous fatty tissue may occur in areas where small atrophic plaques converge. Onychodystrophy and scarring alopecia are typical manifestations.
Skeletal abnormalities present from birth include syndactyly, ectrodactyly, and/or aplasia of the fingers and toes. Additionally, scoliosis, clavicular and costal hypoplasia, and thoracic deformities are commonly observed.
Lateral or complete palatal cleft lip (figure 1) are common congenital conditions. Breaches are typically high and multi-insert. The most frequently observed periodontal sign is gingival dysplasia, which is characterized by a very thin, inflammatory, and occasionally hemorrhagic gum. Generalized marginal gingivitis is also often present [1,3,4].
The most prevalent dental abnormalities include dental dystrophy, microdontia, pointed and/or conical teeth, and less frequently taurodontism and external root resorptions. Other dental issues observed are hypodontia, oligodontia, ectopic and/or delayed eruptions, malocclusions, supernumerary germs, and occasionally fusions and groaning of the temporary teeth. Histologically, the enamel is hypoplastic, which partly accounts for the early onset of caries. The manifestation of these abnormalities varies among patients.
The differential diagnosis is:
- Nevus of Hoffmann-Zurhelle,
- Incontinentia pigmenti
- Congenital
This syndrome, affecting tissues of ectodermal and mesodermal origin, is transmitted in the dominant mode linked to the X chromosome, with lethality in utero for boys, accounting for the increased frequency of miscarriages in these families [3,4 ,5].
The gene responsible is unknown, some authors have described patients with Goltz syndrome and having a deletion of the short arm of the X chromosome with a breakpoint in Xp22.31, suggesting that this may be the location of the gene [4,6].
Only symptomatic dermatological and orthopaedic treatments are possible. Individuals with a very severe form of the syndrome die in infancy, but many patients have a normal life expectancy.
Conclusion
Goltz syndrome is rare, and its oral-maxillofacial signs require an odontologist’s involvement for diagnosis and treatment. Early prevention and regular follow-up are essential to manage dental abnormalities and severe polycavities.
References
1. Wong, HM. “Aetiological factors for developmental defects of enamel”. ‘United States Sports Academy’, 2014
2. Frisk, Sofia. “Studies of genetic mosaicism in rare diseases”. ‘Wiley’, 2022
3. Al-Hadi, Hafidh A, Fox, Keith A, William, Brent. “The Impact of Chronic Liver Diseases on the Level of Heart-Type Fatty Acid Binding Protein (H-FABP) Concentrations”. 2009
4. Bucci, Paolo, Cantile, Tiziana, Caponio, Vito Carlo Alberto, Coppola, et al. “Oral Mucosa and Nails in Genodermatoses: A Diagnostic Challenge”. ‘MDPI AG’, 2021
5. De Vilder, eva, Vanakker, Olivier. “From variome to phenome : pathogenesis, diagnosis and management of ectopic mineralization disorders”. ‘Baishideng Publishing Group Inc.’, 2015
6. Yuen, Wing Yan. “Junctional epidermolysis bullosa”. [s.n.], 2012
7. Dorth, Carroll-Ann Trotman, Hahessy, A. M., Kavanagh, P., McNamara, et al.. “Focal dermal hypoplasia (Goltz-Gorlin) syndrome with taurodontism”. ‘Wiley’, 1996 Kayalvizhi Money, S. “Incidence and Clinicopathological features of Neurocutaneous Disorders.”. 2009
8. Clarke, Angus J., Fete, Mary, Fete, Timothy J., Hadj-Rabia, et al.. “Molecular pathway-based classification of ectodermal dysplasias: first five-yearly update”. ‘MDPI AG’, 2022
This article licensed under the Creative Commons Attribution 4.0 International License CC-BY 4.0., which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are properly credited.